
Aagenaes syndrome is a rare disorder characterized by impaired bile flow from the liver resulting in cholestasis.
Blockage of bile secretion (hepatic cholestasis) results in swelling and fluid retention (lymphedema) in the lower extremities. Neonatal cholestasis in patients with Aagenaes syndrome usually diminishes during early childhood, but remains intermittent in nature. Despite this, Aagenaes syndrome often gradually develops into cirrhosis of liver and giant-cell hepatitis accompanied by scarring of the portal tract tissues.
What is Aagenaes Syndrome?

The disorder is also referred to as lymphedema cholestasis syndrome (LSC1) or cholestasis-lymphedema syndrome (CLS).
Symptoms and diagnosis of Aagenaes syndrome
According to the Human Phenotype Ontology (HPO) database, the most frequent symptoms of Aagenaes syndrome are abdominal pain, swollen legs, obstructive liver disease, abnormality of urine homeostasis, clay-colored (acholic) stools, and fatigue. Other symptoms include enlarged liver, liver scarring, abnormal lipid metabolism, and biliary tract abnormalities.
Unfortunately, there are currently no validated diagnostic tests for the condition. Aagenaes syndrome is diagnosed by evaluation of the symptoms and co-morbidities such as lymphedema.
Prognosis of Aagenaes syndrome
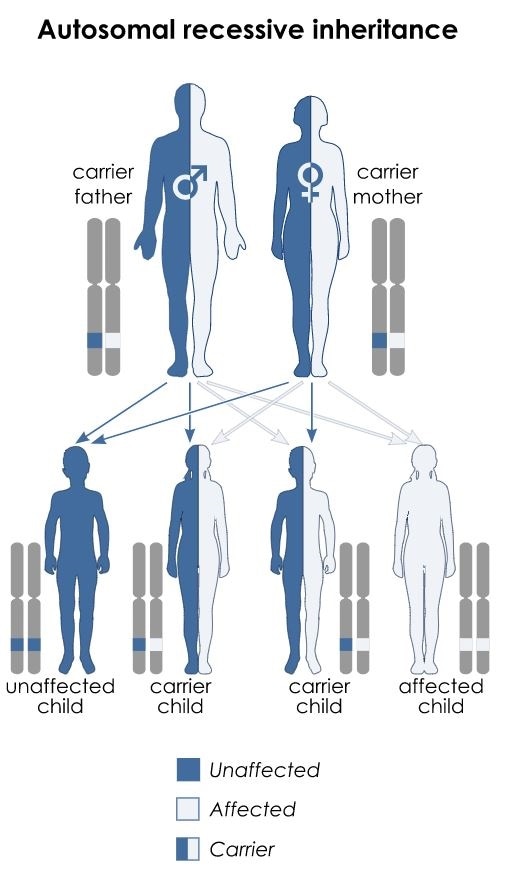
The condition is mainly observed in infants from Norwegian origin, with more than 50% cases being observed in southern Norway. However, it is also prevalent in individuals from other European regions and the United States.The disorder is idiopathic and familial in nature. The exact genetic cause remains inconclusive. However, it is considered to be recessively inherited. The gene is located on chromosome 15q.
Research on Aagenaes syndrome
In a whole-genome screen analysis by Bull et al. (2000), the regions considered similar in the patients with Aagenaes syndrome were identified. The most promising regions were further mapped in larger sets of Norwegian individuals, DNA samples from 8 patients and their 7 non-symptomatic relatives from the same ancestral lineage were evaluated.
The results revealed an extensive similarity in the alleles and haploid genotype of the patients over a region on the chromosome 15q between D15S979 and D15S652 markers, the characteristic which was absent in the unaffected relatives.
This finding suggested that all the Norwegian patients with Aagenaes syndrome could be homozygous with similar gene mutation inherited in dormant form via a common ancestor.
Managing Aagenaes syndrome
There are no established therapeutic options available for curing this disorder. Treatments mainly target specific symptoms especially concerning lymphedema.
Living with a genetic or rare disease can significantly affect the daily lives of patients and their families. As there is a huge scope for further research towards finding a definitive cure for this ailment, community care and support groups are of pivotal importance.
Support and advocacy groups can help patients connect with others with similar condition. These groups provide patient-centred information and are the driving force behind research for better treatments and possible cures. Advisors in these groups help direct patients and their family members to research, resources, and services pertaining to this disorder.
Aagenaes syndrome is named after Oystein Aagenaes, the Norwegian pediatrician who described this syndrome for the first time in 1968. Worldwide, only 100 patients were diagnosed before 2014.
Aagenaes syndrome is also known as Lymphoedema cholestasis syndrome 1. The congenital lymphoid hypoplasia usually affects lymphatic vessels due to intra-hepatic cholestasis. Cholestasis is the pathophysiological condition and develops here as it is associated with the impaired release of bile from the liver and its accumulation within the liver (intra-hepatic).
Progression of cholestasis is one of the primary factors to determine the severity of Aagenaes syndrome. Patients who can survive a prolonged period of cholestasis at their initial stage of the disease can exhibit stable liver disease with better prognosis. More than 50% of Aagenaes syndrome affected individual have a normal lifespan.
By Kashmiri, based on earlier work by Domaina - Own work based on Autosomal dominant - en.svg and Autorecessive.jpg, CC BY-SA 3.0, Link
{kind=link}
{kind=link}
The generalized Aagenaes syndrome symptoms include:
- Abdominal pain
- Light colored stool, which may turn tarry due to intestinal bleeding
- Dark urine
- Enlarged spleen
- Gastrointestinal disturbances
- Fatigue
- Abnormal skin pigmentation
- Pain in joints and bone
Aagenaes Syndrome Symptoms
At the early stage of lifeThe symptoms are gradually progressive and can be classified into two stages of life; early stage and after adolescent age.
- Usually normal birth weight and appetite, but a large amount of fatty, pale colored stools and dark urine are observed.
- Within 2 to 4 weeks of birth, the symptoms of jaundice become prominent, which include yellowing of skin and eye, dark colored urine, extreme weakness, abdominal cramp, nausea, and vomiting.
- The affected infants develop itching which often continues as a skin problem.
- Cholestasis induced malabsorption of fats causes unsatisfactory thrive and growth retardation.
- Deficiency of vitamin A, D, E and K (fat-soluble vitamins) is a common finding and when untreated, increases the risk of rickets, anemia, bleeding tendency and peripheral neuropathy.
- Hemorrhage can cause neonatal death.
- With increasing age, the progressive condition of the disease leads to lymphoedema on the lower limbs due to recurring cholestasis. Children between the ages of 6 - 7 years usually start to develop lymphoedema.
- Lymphoedema is the condition associated with lymphatics hypoplasia, which leads to chronic swelling of the legs. However, depending on the severity of the Aagenaes syndrome, the lymphoedema can extend to arms and periorbital soft tissues present around the eyes, thorax, and gut wall.
- With increasing age, the chronic lymphoedema related symptoms start occurring gradually, such as aching of leg muscles, heaviness in the legs, movement difficulties, and tightening and hardening of skin tissues at the affected region.
- Skin itching starts and often persists as a problem from 6 months of age.
- Although, Aagenaes syndrome affected individuals have normal mental and motor development, impaired learning ability can be observed due to itching or sleepiness.
- Six months to one-year school absenteeism during cholestatic periods may occur in some affected children.
In puberty and in pregnancy
- Cholestasis often causes enlargement of the liver.
- Cholestasis triggers hormonal changes during puberty.
- Increased risk of infection during cholestatic periods.
- An individual suffering from recurrent cholestasis often becomes distressed.
- Results in elevated blood lipid level (Hyperlipidemia).
- Some Aagenaes syndrome affected adolescents may develop lymphoedema before puberty.
- Aagenaes syndrome-affected individuals face several social implications due to lymphoedema and recurrent bacterial skin infections like erysipelas, which causes red patches on the skin.
- Severe lymphoedema in adults can become a reason for disability.
- Portal hypertension can cause oesophageal variceal bleeding due to enlargement of the vein in the oesophagus, fluid accumulation in the peritoneal cavity and over-activity of the spleen.
- Recurrent cholestasis may cause liver cirrhosis in adults.
- Aagenaes syndrome in pregnant women increases the risk of premature delivery and stillbirth due to cholestasis.
How is Aagenaes Syndrome Diagnosed?
Aagenaes syndrome is a rare disease, and the only identified hereditary lymphoedema which slows down or ceases bile flow resulting in cholestasis. However, lymphoedema may develop on its own due to other underlying hepatic ailments or lymphatic system disorders.
Aagenaes syndrome can easily be misdiagnosed with biliary atresia, leading to unnecessary surgical intervention. Therefore, a proper diagnosis is imperative to start a correct clinical treatment of Aagenaes syndrome.
The diagnosis of Aagenaes syndrome is challenging because the early signs of lymphoedema are difficult to detect, which further delays initiation of medical intervention to treat Aagenaes syndrome.
If someone is affected by Aagenaes syndrome first in the family then it becomes difficult to diagnose until the lymphoedema symptom appears. Cholestasis can be diagnosed within the first year of age, but in some patients with Aagenaes syndrome, lymphoedema develops gradually with age.
Diagnosis based on clinical evaluation
Jaundice, dark colored urine, pale colored stool, fatigue, and pruritus are the common clinical signs for both intra and extra-hepatic cholestasis, which makes it difficult to determine the actual underlying cause.
The existence of Aagenaes syndrome in family history can be considered as a major finding in the clinical investigation of this disease. Clinical diagnosis of lymphoedema at the initial stage is the most challenging until the symptoms appear which further delays the confirmation of Aagenaes syndrome.A thorough assessment of patient history and careful physical examination are fundamental to diagnose Aagenaes syndrome.
Depending upon the onset, lymphoedema can be divided into three categories like congenital, praecox, or tarda. In case of congenital lymphoedema, the symptoms usually appear within 2 years of age.
In praecox lymphoedema, the onset of symptoms start inbetween 2 to 35 years; however, in most of the cases, it appears near puberty. Tarda is a type of inherent lymphoedema, which usually occurs after 35 years of age. Rates of dyslipidemia and portal hypertension are also common clinical findings in Aagenaes syndrome.
Biomarkers for Aagenaes Syndrome
Detection of jaundice is usually followed by identification of certain biomarkers which are diagnosed through blood tests. Within one month of birth, the child with Aagenaes syndrome gets elevated levels of serum bilirubin concentration which may gradually normalize after 3 to 4 years of age.
An increased level of bile acid, i.e. more than 40 μmol/L within 6 months from birth, is observed, which gradually lowers to an extent and reaches a moderately high level within 6 years of age.
Aspartate transaminase (ALT) and alanine transaminase (AST) are two liver enzymes of which levels become extremely elevated in the blood within one month from birth; levels then drop to moderate levels at 2 to 3 years of age.
The upper limit of Gamma-glutamyl transpeptidase level is almost twice the normal limit. The use of phenobarbital may further increase Gamma-glutamyl transpeptidase level specifically in patients with Aagenaes syndrome.
Instrumental investigations
There are some essential investigations to assess the hepatic conditions:
Abdominal ultrasonography and Magnetic resonance cholangiopancreatography (MRCP) are two techniques used to diagnose Aagenaes syndrome. However, in some patients, the results of these tests can be unclear. In such cases, the patients serum is analyzed for the presence of antimitochondrial antibodies (AMA) or a liver biopsy is taken,
Lymphoscintigraphy is used to determine lymphatic anatomy. The presence of lymphoedema is determined using following methods:
- Lymphoscintigraphy
- Computed tomography
- Magnetic resonance imaging
- Duplex ultrasound
Genetic Findings
The precise genetic mutation which causes Aagenaes Syndrome has not yet been elucidated. An autosomal recessive inheritance pattern is shown in Aagenaes Syndrome, and the long arm of chromosome 15q has been attributed to the genetic cause in some cases, but this has not been proven in all cases.
References:
- https://rarediseases.info.nih.gov/diseases/370/aagenaes-syndrome
- http://www.checkorphan.org/diseases/aagenaes-syndrome
- https://www.sciencedirect.com/science/article/pii/S0002929707632935
- http://www.lymphedemapeople.com/thesite/Aagenaes_syndrome.html
- https://www.medigest.uk/diseases/aagenaes-syndrome/
- https://rarediseases.info.nih.gov/diseases/370/aagenaes-syndrome
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3734684/
0Comments